*NURSING > QUESTIONS & ANSWERS > NURS 6501 Knowledge Check Week 11 (answered & graded) latest Spring 2021 (All)
NURS 6501 Knowledge Check Week 11 (answered & graded) latest Spring 2021
Document Content and Description Below
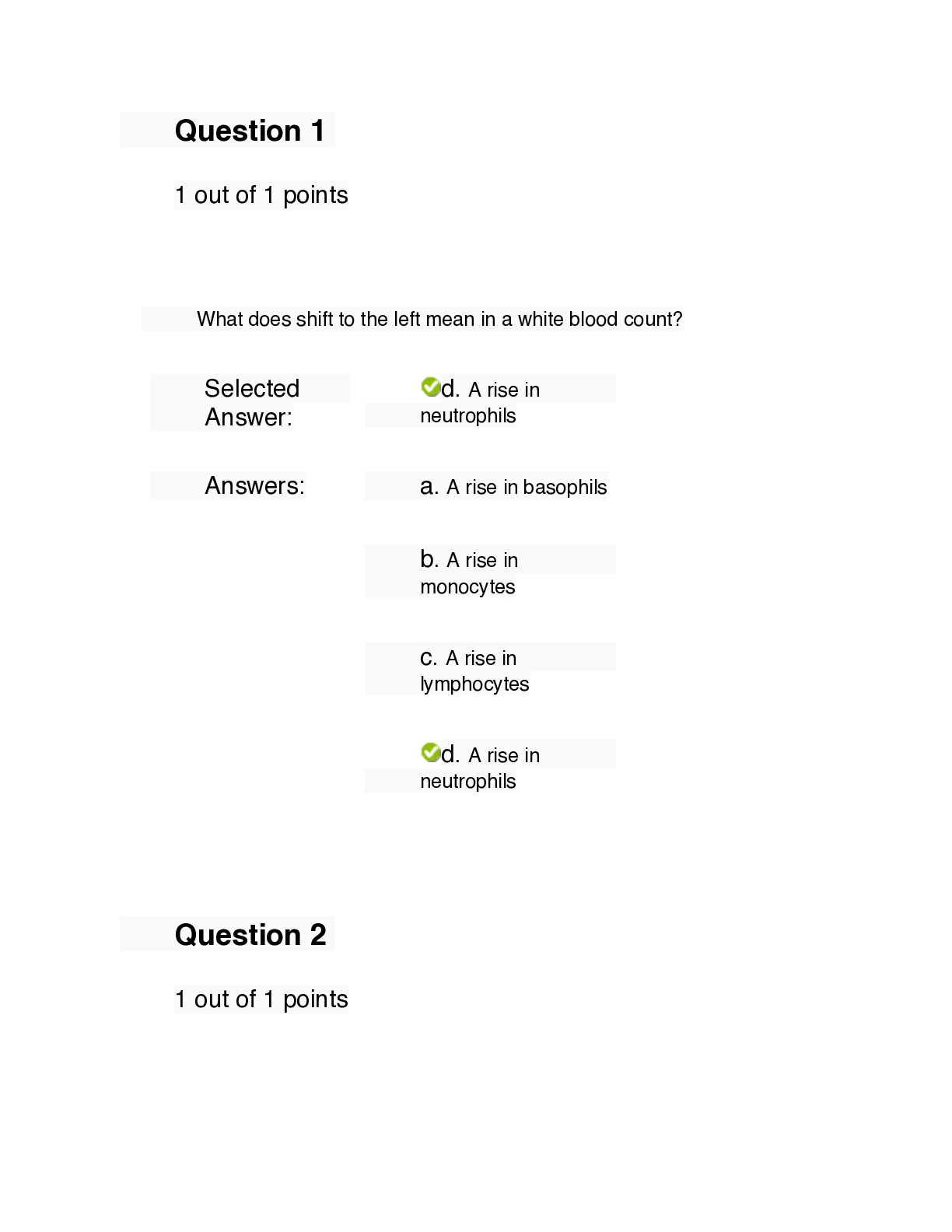
NURS 6501 Knowledge Check Week 11 (answered & graded) A ten-year-old boy is brought to clinic by his mother who states that the boy has been listless and not eating. She also notes that he has been e... asily bruising without trauma as he says he is too tired to go out and play. He says his bones hurt sometimes. Mother states the child has had intermittent fevers that respond to acetaminophen. Maternal history negative for pre, intra, or post-partum problems. Child’s past medical history negative and he easily reached developmental milestones. Physical exam reveals a thin, very pale child who has bruises on his arms and legs in no particular pattern. The APRN orders complete blood count (CBC), and complete metabolic profile (CMP). The CBC revealed Hemoglobin of 6.9/dl, hematocrit of 19%, and platelet count of 80,000/mm3. The CMP demonstrated a blood urea nitrogen (BUN) of 34m g/dl and creatinine of 2.9 mg/dl. The APRN recognizes that the patient appears to have acute leukemia and renal failure and immediately refers the patient to the Emergency Room where a pediatric hematologist has been consulted and is waiting for the boy and his mother. The diagnosis of acute lymphoblastic leukemia (ALL) was made after extensive testing. Question 1 of 2: What is ALL? Acute lymphoblastic leukemia (ALL) is a cancer of the blood-forming tissues, such as the bone marrow. Leukemia most often produces abnormal white blood cells called leukemic cells. Once in the blood, leukemic cells can spread to other organs, such as the lymph nodes, spleen, and brain. ALL is composed of immature B (pre-B) or T (pre-T) cells called lymphoblasts. The bone marrow is dense with lymphoblasts, considered hypercellular, that replace the normal marrow and disrupt normal function. Many of the chromosomal abnormalities documented in ALL cause dysregulation of the expression and function of transcription factors required for normal B-cell and T-cell development. The mutations can include both gains of function and loss of function that are required for normal development. ALL make up 75% to 80% of all childhood leukemias. Question 2 of 2: How does renal failure occur in some patients with ALL? Renal failure can occur in some individuals with ALL as a result of high uric acid levels (hyperuremia). This is more common at diagnosis or during active treatment. Uric acid levels rise as an end product of purine metabolism from cellular destruction. Because the major pathway of excretion is through the kidneys, urates can precipitate in the renal tubules or ureters and can lead to low urine output (oliguria) and acute renal failure. Renal failure is preventable if uric acid levels are monitored and treatment is aimed at optimal hydration. Question 3: A 12-year-old female with known sickle cell disease (SCD) present to the Emergency Room in sickle cell crisis. The patient is crying with pain and states this is the third acute episode she has had in the last nine months. Both parents are present and appear very anxious and teary eyed. A diagnosis of acute sickle cell crisis was made. Appropriate therapeutic interventions were initiated by the APRN and the patient’s pain level decreased, and she was transferred to the pediatric intensive care unit (PICU) for observation and further management. Question 1 of 2 What is the pathophysiology of acute SCD crisis and why is pain the predominant feature of acute crises? The pathogenesis of sickling includes erythrocyte derangement, chronic hemolysis, microvascular occlusions, and tissue damage. Deoxygenation is probably the most important variable in determining the occurrence of sickling. Other significant variables that affect sickling include the interaction of Hbs with other types of hemoglobin in the cell, mean cell hemoglobin concentration, intracellular pH, and transit times of erythrocytes through the microcirculation. The pain associated with sickle cell disease is a result of hypoxic injury and infarction to areas of the body including bones, lungs, spleen, liver, and penis. Bone pain is very common in children with sickle cell disease as a result of the hypoxic injury. Question 4: Discuss the genetic basis for SCD. Sickle cell disease (SCD) is a common hereditary hemoglobinopathy where HbS is formed by a genetic point mutation (missense) in B-globulin that leads to the replacement of one glutamate amino acid with a valine amino acid. Abnormal versions of B-globin can distort erythrocytes into a sickle shape. Hemoglobin consists of four protein subunits called alpha-globin and two subunits called B-globin. The hemoglobin B (HbB) gene provides instructions for making protein B-globin. Other mutations in the HbB gene lead to other versions of B-globulin such as hemoglobin C (HbC) and hemoglobin E (HbE). HbB gene mutations can also affect the quantity of B-globulin, such as low levels of B-globin found in B-thalassemia. SCD denotes all genotypes containing at least one sickle gene, in which HbS makes up at least half the hemoglobin present. The most prevalent SCD genotypes include homozygous hemoglobin SS and the compound heterozygous conditions hemoglobin SB0-thalassemia (HbB0-thalassemia), hemoglobin SB-thalassemia (HbSB+-thalassemia), and hemoglobin SC disease (HbSC). HbSS and HbSB0-thalassemia are clinically similar and are commonly referred to as sickle cell anemia. These genotypes are associated with the most severe clinical manifestations. People who inherit two genes for sickle hemoglobin (one from each parent) have sickle cell disease. With a few exceptions, a child can inherit sickle cell disease only if both parents have one gene for sickle cell hemoglobin. Question 5: The parents of a 9-month boy bring the infant to the pediatrician’s office for evaluation of a swollen right knee and excessive bruising. The parents have noticed that the baby began having bruising about a month ago but thought the bruising was due to the child’s attempts to crawl. They became concerned when the baby woke up with a swollen knee. Infant up to date on all immunizations, has not had any medical problems since birth and has met all developmental milestones. Pre-natal, intra-natal, and post-natal history of mother noncontributory. Family history negative for any history of bleeding disorders or other major genetic diseases. Physical exam within normal limits except for obvious bruising on the extremities and right knee. Knee is swollen but no warmth appreciated. Range of motion of knee limited due to the swelling. The pediatrician suspects the child has hemophilia and orders a full bleeding panel workup which confirms the diagnosis of hemophilia A. Question 1 of 2: Explain the genetics of hemophilia. The major types of hemophilia are hemophilia A and hemophilia B. Type A is the most common. Hemophilia A is caused by changes in the F8 gene and hemophilia B is caused by mutations in the F9 gene. The F8 gene provides instructions for making the protein called coagulation VIII. F9 produces coagulation factor IX. These factors are important for blood clotting. Factor VIII is an essential cofactor for factor IX in the coagulation cascade. Alterations in coagulation factors decrease the ability to form clots in response to injury. Decreased blood clotting leads to continuous bleeding. Hemophilia A and B are inherited as an X-linked recessive pattern. The genes associated with these disorders are located on the X-chromosome; in males, one altered copy of the disorder causes the condition, which is why it affects mainly males (females are carriers) and very rarely homozygous females. In approximately 30% of individuals with no family history, their disease is caused by acquired new mutations. The degree of severity of hemophilia disease is dependent on the concentrations of clotting factor VIII or IX in the blood. Severe hemophilia being a result of concentrations of clotting factors of 1% or less in the blood. Question 6: Briefly describe the pathophysiology of Hemophilia. Multiple types of mutations are associated with both types of hemophilia. Hemophilia A is described with more than 2000 unique mutations and B has more than 1000 described mutations. Inversions in introns 1 and 22 of the factor VIII gene are the most frequently observed mutations and account for the majority of severe cases of hemophilia. Inadequate factor VIII results in the insufficient generation of thrombin by the FIXa and FVIIIa complex through the intrinsic pathway of the coagulation cascade. Point mutations, in which a single base in the DNA is inserted in the place of another base, are another type of mutation that causes hemophilia. Point mutations comprise the majority of unique mutations associated with hemophilia. When a point mutation gives rise to a de novo stoop codon, translation of the protein ceases and a shortened version of the protein is synthesized. This type of defect also is associated with severe hemophilia, with coagulant activity levels below 1%. Resulting in severe, spontaneous bleeding. The altered amino acid chain can destroy protein function, activation, or folding; inhibit intracellular processing, or cause protein clearance. Question 7: During a routine 16-week pre-natal ultrasound, spina bifida with myelomeningocele was detected in the fetus. The parents continued the pregnancy and labor was induced at 38 weeks with the birth of a female infant with an obvious defect at Lumbar Level 2. The Apgar Score was 7 and 9. The infant was otherwise healthy. The sac was leaking cerebral spinal fluid and the child was immediately taken to the operating room for coverage of the open sac. The infant remained in the neonatal intensive care unit (NICU) for several weeks then discharged home with the parents after a prescribed treatment plan was developed and the parents were educated on how to care for this infant. Question 1 of 2: What is the underlying pathophysiology of myelomeningocele? Myelomeningocele is a hernial protrusion of a saclike cyst (containing meninges, spinal fluid, and a portion of the spinal cord with its nerves) through a defect in the posterior arch of a vertebra. It occurs when a developing fetus's spinal cord fails to fully develop or close properly while in-utero for which there is no cure. The spinal canal is open along several vertebrae in the lower or middle back. 80% of myelomeningoceles are located in the lumbar and lumbosacral regions, the last of the neural tube to close. The anatomic level of the myelomeningocele sac correlates with the patient's neurologic, motor, and sensory deficits. The spinal cord and nerve roots are malformed below the level of the lesion, resulting in loss of motor, sensory, reflex, and autonomic functions. Question 8: Describe the pathophysiology of hydrocephalus in infants with myelomeningocele. Hydrocephalus occurs in 85% of infants with myelomeningocele. Myelomeningoceles are almost always associated with a brain malformation called a type II Arnold-Chiari malformation. This a complex malformation of the brainstem and cerebellum in which the cerebellar tonsils are displaced downward into the cervical spinal canal, the upper medulla and lower pons are elongated and thin and the medulla also is displaced downward and sometimes has a “kink”. This malformation is associated with hydrocephalus from the pressure that blocks the flow of CSF and syringomyelia, and abnormality causing cysts at multiple levels within the spinal canal. Question 9: A preterm infant was delivered at 32 weeks gestation and was taken to the NICU for critical care management. Physical assessment of the chest and heart remarkable for a continuous-machinery type murmur best heard at the left upper sternal border through systole and diastole. The infant had bounding pulses, an active precordium, and a palpable thrill. The infant was diagnosed with a patent ductus arteriosus (PDA). Discuss the hemodynamic consequences of a PDA. The patent ductus arteriosus (PDA) is a vessel located between the junction of the main and left pulmonary arteries and lesser curvature of the descending aorta. After birth, a decrease of prostacyclins and prostaglandin concentration usually causes arterial duct closure. Failure of the PDA to close results in persistent patency of the ductus arteriosus. The hemodynamic effects of PDA depend on the size of the lumen and the resistance in the pulmonary and systemic circulations. Pulmonary vascular resistance (PVR) and systemic vascular resistance (SVR) are almost equal at birth and are reflected in the pulmonary artery (PA) and aorta, respectively; therefore shunting is minimal. However, as PVR drops, a reversal of fetal shunting occurs. Blood now begins to shunt left to right from the aorta to the PA. The hemodynamic effect is increased pulmonary blood flow, resulting in increased pulmonary venous return to the left atrium (LA) and left ventricle (LV) with an increased workload on the left side of the heart. The increased workload is caused by the increased pulmonary venous return to the LA and potentially an increase in right ventricular pressure if pulmonary vascular changes occur in response to the increased blood flow leading to an increase in pulmonary vascular pressure. This results in hypoperfusion to organs, vascular congestion, and pulmonary edema, and leads to heart failure if not corrected. Question 10: A 7-year-old male was referred to the school psychologist for disruptive behavior in the classroom. The parents told the psychologist that the boy has been difficult to manage at home as well. His scholastic work has gotten worse over the last 6 months and he is not meeting educational benchmarks. His parents are also worried that he isn’t growing like the other kids in the neighborhood. He has been bullied by other children which are contributing to his behaviors. The psychologist suggests that the parents have some blood work done to check for any abnormalities. The complete blood count (CBC) revealed hypochromic microcytic anemia. Further testing revealed the child had a venous lead level of 21 mcg/dl (normal is < 10 mcg/dl). The child was diagnosed with lead poisoning and it was discovered he lived in public housing that had not finished stripping lead paint from the walls and woodwork. How does lead poisoning account for the child’s symptoms? Lead poisoning results in high blood lead levels. No level of lead in the blood is safe. The neurological system is most vulnerable to lead toxicity. The neurological symptoms related to lead poisoning occurs as lead inhibits sulfhydryl enzymes, which leads to increased membrane permeability, followed by an increase in intracranial pressure and cerebral edema, leading to tissue ischemia, followed by irreversible necrosis and tissue atrophy, resulting in behavior changes, lowered IQ, intellectual disability, convulsions, coma, and in severe cases or if left untreated, death. Skeletal system symptoms (impaired growth) are a result of accumulated tertiary lead phosphate and calcium, which interferes with the growth of metaphyses in long bones, and leads to a decrease growth rate. Anemia results from impaired uptake and utilization of iron and the prevention of the formation of hemoglobin as a result of lead in the bloodstream. Question 11: Emergency Medical Services (EMS) was dispatched to a home to evaluate the report of an unresponsive 3-month-old infant. Upon arrival, the EMS found a frantic attempt by the presumed father to resuscitate an infant. The EMS took over and attempted CPR but was unable to restore pulse or respiration. The infant was transported to the Emergency Room where the physician pronounced the child died of Sudden Infant Death Syndrome (SIDS). The distraught parents were questioned as to the events surrounding the discovery of the baby. Parents state the child was in good health, had taken a full 6-ounce bottle of formula before being put down for the evening. The child had been sleeping through the night before this. Parents stated the baby had had some “sniffles” a few days before and was taken to the pediatrician who diagnosed the child with a mild upper respiratory tract viral syndrome. No other pertinent history. What is thought to be the underlying pathophysiology of SIDS? The etiology of SIDS remains unknown but probably involves a combination of predisposing factors. These include a vulnerable infant in a critical developmental period for homeostatic control with altered cardiorespiratory, circulatory, and arousal characteristics, and environmental stressors (i.e. prone or side-resting position, soft bedding, or in utero or environmental tobacco exposure). Other theories involve immune dysregulation, airway inflammation, and responses to either bacterial pathogens from the nasopharynx or viral respiratory tract infections. There is growing evidence that genetic factors may predispose certain individuals to SIDS. The most important risk factor genes include those involved in the regulation of the immune system and inflammation, cardiac function, and brainstem function. SIDS almost always occurs during nighttime sleep, when infants are least likely to be observed. A seasonal variation has been noted, which higher frequencies during the winter months. This has been related to a higher rate of respiratory tract infection during those months, and such infections are often reported to have preceded death. QUESTION 12 A 4-year-old female is brought to the pediatrician by her mother who states the child has been running a fever to 102.0 F, has a “pink eye”, and that her tongue looks very bright red and swollen. The mother states the fever has been present for 5 days, noticed the child had developed a rash and that the child’s legs look “puffy”. No other symptoms were noted. Past medical history noncontributory. All immunizations up to date. Physical exam remarkable for current fever of 102.8 F, bilateral conjunctivitis without purulent material, oral mucosa with bright red erythema, dry, with fissuring of the lips. Legs are noted to have peripheral edema and are also erythematous. Palmar desquamation noted. There are fine maculopapular rash and + cervical adenopathy. The presumptive diagnosis currently (pending laboratory data) is Kawasaki Disease. Question 1 of 2: What is Kawasaki Disease and what is the pathophysiology? Kawasaki disease (KD), also known as mucocutaneous lymph node syndrome and Kawasaki syndrome, is an acute, self-limiting systemic vasculitis that may result in cardiac sequelae. KD occurs throughout the world with most cases occurring in Japan. The etiology of KD remains unknown, however, theories center on immunologic response to an infectious, toxic, or antigenic substance. KD progresses pathologically and clinically in the following stages: Stage 1 (days 1-12)- Small capillaries, arterioles, and venules become inflamed, as does the heart itself. Stage 2 (days 13-25)- Inflammation spreads to larger vessels, and aneurysms of the coronary arteries develop. Stage 3 (days 26-40)- Medium-size arteries begin the granulation process, causing coronary artery thickening; inflammation resolves in the microcirculation; there is a risk of thrombus formation. Stage 4 (day 41 and beyond)- The vessel wall eventually develops scarring, intimal thickening, calcification, and stenosis of coronary arteries. Question 13: How does Kawasaki Disease cause coronary aneurysms? Small capillaries, arterioles, and venules become inflamed along with the heart itself. As inflammation spreads to larger vessels this leads to the formation of coronary artery aneurysms. Aneurysm result as inflammation of the vessels weakens the vessel walls and as the pressure in the arteries is normally high, this pressure can lead to outpouchings of the vessel wall resulting in an aneurysm. QUESTION 14: A 9-year-old boy was brought to the Urgent Care Center by his parents who state that the child had a sudden onset of difficulty catching his breath, has a new cough and is making a “funny sound” when he breathes. The parents state there is no prior history of this, and the child had not been ill prior to the start of the symptoms. Past medical history noncontributory. No family history of respiratory problems. No known allergies to drugs or food. Physical exam positive for respiratory rate of 26, use of accessory muscles, with suprasternal retractions, heart rate of 132 beats per minute, an audible inspiratory and expiratory wheeze noted, and the pulse oximetry is 89% on room air. After the APRN institutes appropriate urgent treatment, the child’s breathing slowly returned to normal, vital signs normalize, and the pulse oximetry increases to 97%. The APRN suspects the child has asthma and tells the parents that they need to bring the child to a pulmonologist for further evaluation and care. What is the underlying pathophysiology of asthma? Asthma is a chronic inflammatory disease characterized by bronchial hyperreactivity and reversible airflow obstruction, usually caused by an allergen. Airway epithelial exposure to antigen initiates both an innate and adaptive immune response in sensitized individuals. Many cells and cellular elements contribute to the persistent inflammation of the bronchial mucosa and hyperresponsiveness of the airways, including dendritic cells, T helper 2 lymphocytes, B lymphocytes, mast cells, neutrophils, eosinophils, and basophils. In the early asthmatic response phase, antigen exposure in the bronchial mucosa activates dendritic cells that bring the antigen to the T helper cells. As a result, many inflammatory cytokines are released. This causes increased capillary permeability, mucosal edema, bronchospasm, and tenacious mucus secretion from mucosal goblet cells with narrowing of the airways and obstruction to airflow. In the second phase (approximately 4-8 hours after the early asthmatic response). This leads to an increase in airway hyperresponsiveness. This causes chemotactic recruitment of lymphocyte, eosinophils, neutrophils, basophils, and lymphocytes during the acute response and causes a latent release of inflammatory mediators and newly formed mediators. These combined mediators result in bronchospasm, edema, and mucus secretion with airflow obstruction. Airway obstruction increases resistance to airflow and decreases flow rates especially expiratory flow. Impaired expiration causes air trapping, hyperinflation distal to obstructions, and increased work of breathing. Hyperinflation compensates for the airflow obstruction, but this compensation is limited when the tidal volume approaches the volume of the pulmonary dead space; the result is alveolar hypoventilation. Uneven changes in airflow resistance result in uneven distribution of air and modifications in circulation from increased intra-alveolar pressure due to hyperinflation leading to ventilation-perfusion mismatch. QUESTION 15: A 24-year-old female with known cystic fibrosis (CF) has been admitted to the hospital for evaluation for a possible lung transplant. She was diagnosed with CF when she was 9 months old and has had multiple hospitalizations for pneumonia, respiratory failure, and small bowel obstructions. She currently is oxygen dependent and has been told by her physicians that she has end-stage pulmonary disease secondary to CF. The only recourse for her currently is a lung transplant. Question 1 of 2: What is cystic fibrosis and discuss the pathophysiology Cystic fibrosis (CF) is an autosomal recessive inherited disease that results from defective epithelial chloride ion transport. The CF gene is located on chromosome 7. CF is a multiorgan disease that affects the airways, digestive tract, and reproductive organs. The cystic fibrosis transmembrane conductance regulator (CFTCR) gene mutation results in the abnormal expression of cystic fibrosis transmembrane conductance regulator (CFTCR) protein. The CFTCR protein is an activated chloride channel present on the surface of many different epithelial cells, including those in the airways, bile ducts, the pancreas, sweat ducts, paranasal sinuses, and the vas deferens. The effect on the lungs is the most important and respiratory failure is the most common cause of death. Features of CF lung disease are mucus plugging, chronic inflammation, and chronic infection of the small airways. CF mucus is dehydrated and viscous because of defective chloride secretion and excess sodium absorption and ion transport. Leading to increased water absorption & impaired ciliary function, followed by impaired mucus clearance and airway obstruction, which allows bacteria to adhere to the epithelium, followed by excessive increases in neutrophils leading to damage of the lungs from oxidants and protease release, this, in turn, causes chronic bacterial infections, followed by bronchiectasis, and ultimately respiratory failure. Question 16: Question 2 of 2: What is the reason people with CF are often malnourished? Nutritional problems are extremely common in CF and poor nutrition is correlated with worse outcomes along with additional complications such as decreased bone miner density, and growth failure. Approximately 90% of CF children have pancreatic insufficiency as a result of abnormal ion transport causing decreased fluid and bicarbonate secretion from the pancreatic acinar cells, leading to thickened secretions plugging the smaller pancreatic ducts and eventual autodigestion or atrophy of the acinar cells. This causes fat malabsorption which causes diminished absorption of fat-soluble vitamins (i.e. A, D, E, & K). This causes reduced nutrient absorption and leads to growth failure, poor clinical outcomes, and reduced life expectancy. Individuals must take exogenous pancreatic enzymes with meals and snacks to absorb nutrients and control malabsorptive symptoms. QUESTION 17 A 14-year old girl who was trying out for cheerleading underwent a physical examination by the APRN who notices that the girl had uneven hip height, asymmetry of the shoulder height, shoulder and scapular prominence, and rib prominence. The rest of the physical exam was normal and the APRN referred the girl to an orthopaedist for evaluation for possible scoliosis. Radiographs in the orthopedic office confirm the diagnosis of idiopathic scoliosis. The spinal curve was measured at 26 degrees and it was recommended that the girl be fit for a low-profile back brace. What is thought to be the pathophysiology of idiopathic scoliosis? Idiopathic scoliosis is classified as infantile, juvenile, or adolescent, depending on the child's age at the time of onset. The exact cause of scoliosis is still unknown. However, in individuals with scoliosis, it has been hypothesized that there is an abnormality of the central nervous system involving the balance mechanism (reticular system) in the midbrain. A genetic component has also been suggested as 30% of cases occur in families. Also, individuals with adolescent idiopathic scoliosis have an abnormality in the function of the posterior columns of the spinal cord. This results in abnormal proprioception and is not evident clinically except in the presence of scoliosis. The earliest pathologic changes occur in the soft tissues. The muscles, ligaments, and other soft tissues become shortened on the concave side of the curve. The vertebral deformity occurs as asymmetric forces are applied to the epiphyseal center of the ossification by shortened and tight soft tissue on the concave side of the curve. True curves involve not only bending but also twisting of the torso, leading to a rib hump seen when the child leans forward. The curve increases most rapidly during periods of rapid skeletal growth. If the curve is less than 40 degrees at skeletal maturity, the risk of progression is small. In curves greater than 50 degrees, the spine is biomechanically unstable, and the curve usually progresses even after the cessation of growth. Curves in the thoracic spine over 80 degrees result in decreased pulmonary function. Back pain is the most common symptom of large curves. QUESTION 18 A 2-year-old boy was brought to Urgent Care by his parents who state the boy has been having large amounts of diarrhea, been very irritable, and very pale. The parents noticed there was blood in diarrhea and when the boy’s legs became swollen, they sought care. Past medical history noncontributory and all immunizations up to date. Social history noncontributory and the child is in daycare 5 days a week. No known exposure to other sick children and the only new event the parents could think of is the daycare workers took the children to a local petting zoo about a week ago. Physical exam revealed a pale, ill-appearing child with swollen legs, tender abdomen, and petechia on the legs and abdomen. The APRN suspects the child may have been exposed to a bacterium at the petting zoo and arranges for the patient to be transferred to the Emergency Room. There the child was found to be in renal failure, have hypertension, and was diagnosed with hemolytic uremic syndrome (HUS). What is the pathophysiology of HUS? Hemolytic uremic syndrome (HUS) is an acute disorder characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal impairment. HUS has been associated with both bacterial and viral agents, as well as endotoxins, especially from E. Coli 0157:H7 and recently from E. Coli 0104:H4. Potential sources of exposure include animals, unpasteurized beverages, and contaminated meat and vegetables. In HUS, verotoxin (Shiga toxin) from E. coli is absorbed from the intestines into the blood binds to polymorphonuclear leukocytes, and is transported to the kidney, causing a cascade of effect, including lysis of glomerular capillary endothelial cells, separation of endothelial cells from the basement membrane, activation, and aggregation of platelets and activation of the coagulation cascade. The glomerular arterioles become swollen and occluded with platelets and fibrin clots. There are decreased glomerular filtration and the damaged glomerular membrane results in hematuria and proteinuria. Narrow vessels injure passing erythrocytes. The damaged erythrocytes are removed by the spleen, causing acute hemolytic anemia. The platelet clustering within damaged vessels, combined with the damage and removal of platelets produces thrombocytopenia. Fibrin-rich thrombi can be found throughout the microcirculation. QUESTION 19 The parents of a 3-year-old boy bring the child to the pediatrician with concerns that their child seems “small for his age”. The parents state that the boy has always been small but did not worry until the child went to day care and they noticed other children of the same age were much bigger. They also note that his teeth were very late in coming in. Normal prenatal, perinatal and postnatal history and no medical history on either side of family regarding issues with growth and development. Physical exam is normal except for short limbs and small teeth. The pediatrician suspects the child has pituitary dwarfism. A complete laboratory and radiographic work up confirmed the diagnosis. What is the pathophysiology of pituitary dwarfism? Pituitary dwarfism is a result of a deficiency in growth hormone (GH). It occurs in children and adults. Children with GH deficiency have abnormally short stature with normal body proportions. GH deficiency can result from any causes of hypopituitarism. Several genetic defects have been identified in the GH axis in children, including a recessive mutation in the GH gene, resulting in failure of GH secretion. These include a recessive mutation in the GHRH gene, failing GH secretion, and mutations that cause GH insensitivity by affecting the GH receptor, IFG-1 biosynthesis, IGF-1 biosynthesis, IGF-1 receptors, or defects in GH signal transduction. GH in children is manifested by growth failure. Another feature of GH in children are fasting hypoglycemia as a result of impaired substrate mobilization for gluconeogenesis and enhanced insulin sensitivity. Question 20: A 4-year-old boy was brought to the Emergency Room by his parents with a suspected femur fracture. The parents state the child was playing on the couch when he rolled off and cried out in pain. There were no other injuries noted. A review of the child’s chart revealed this was the 4th Emergency Room visit in the last 15 months for fractures after low impact injury. The parents were suspected of child abuse and Child and Protective Services were consulted. The APRN assessing the child noted that the child had unusually thin and translucent skin, poor dentition, and blue sclera. The APRN suspects the child may have osteogenesis imperfecta (OI). Laboratory results revealed an elevated serum alkaline phosphatase and the diagnosis OI was made based on the clinical picture and elevated alkaline phosphatase. What is the pathophysiology of OI? The major errors in OI lie in the synthesis of collagen, a triple helix with two matching alpha chains, and one beta chain. Collagen is presented in the bone, cartilage, eye tissue, skin, and vascular system. The severity of the OI phenotype and the related anomalies of the eye, dentition, or vascular system are all dependent on the severity of the genetic anomaly and the part of the triple helix that is affected. Several metabolic abnormalities are associated with OI. Some individuals have increased serum thyroxine levels, suggesting hyperthyroidism. This is consistent and coincides with the findings of increased sweating, heat intolerance, increased body temperature, resting tachycardia, and tachypnea found in individuals with hyperthyroidism. [Show More]
Last updated: 1 year ago
Preview 1 out of 11 pages

Reviews( 0 )
Document information
Connected school, study & course
About the document
Uploaded On
Feb 07, 2021
Number of pages
11
Written in

Additional information
This document has been written for:
Uploaded
Feb 07, 2021
Downloads
0
Views
52
.png)
.png)





















.png)


.png)


.png)


